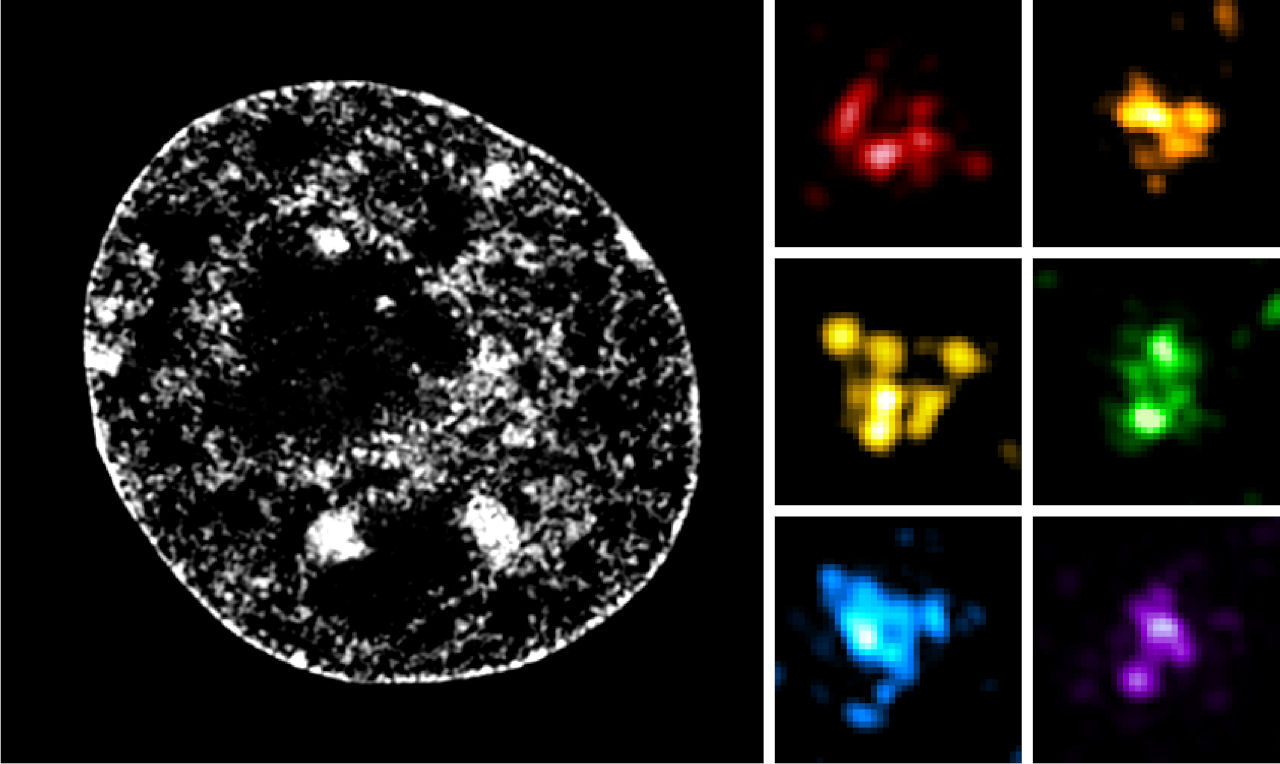
3D organization and function of the genome
The information stored in our genome is intertwined with its function, such that, when cells are submitted to specific sets of conditions, they may pass on to their progeny their functional state. Since DNA has been identified as a critical carrier of genetic information and since the same DNA can correspond to alternative, heritable functional states in certain cases, this transmission of cellular memory has been dubbed epigenetic inheritance. In the most spectacular way, this extends to inheritance of a phenotypic trait into subsequent generation, a phenomenon for which Conrad H. Waddington provided evidence more than seventy years ago and which is well documented in plants. However, to which extent epigenetic inheritance operates in animals is hotly debated. Chromatin and its higher‑order organization are epigenetic components that play an essential role in genome regulation. Both the DNA molecule and the nucleosomal histones can be extensively modified in a way that impinges on gene expression and may be inherited as well as erased upon specific regulatory cues. Furthermore, chromatin fibers can be folded into yet higher‑order structures and chromosomes are confined in discrete “territories”.
We and others have discovered that metazoan chromosomes share a modular organization of their chromatin in structures called “Physical domains” or “Topologically Associating Domains” (TADs). TADs can be defined as linear units of chromatin that fold as discrete three‑dimensional (3D) structures tending to favor internal, rather than external, chromatin interactions. TADs are delimited by boundaries, which contain housekeeping genes and insulator sites. They are detected by methods such as Hi‑C, which allows genome‑wide identification of chromatin contacts, and they correspond to Chromosomal Domains (CDs), previously identified by microscopy. The investigation of chromatin landscapes in metazoa through genome‑wide association studies proved to be a fruitful approach. Theoretically, a huge number of chromatin types based on different combinations of chromatin‑associated marks would be possible but, in fact, every report basically recapitulated the presence of an active chromatin environment, that can be further subdivided because of specific differences between gene promoters, coding regions and distal regulatory elements (enhancers), and of three major types of repressive chromatin: a Polycomb‑repressed environment, a heterochromatic environment and a so-called null environment that is repressed without strong active silencing mechanisms. Strikingly, TADs were found to overlap with linear chromatin domains, indicating that epigenomic labeling of chromosome domains is intimately linked to their 3D folding.
We are trying to understand the principles governing 3D folding of the genome, from establishment of chromatin loops to the generation of chromosome domains, compartments, territories and the establishment of interchromosomal interactions. We use Drosophila , but also mouse and human cells, and state of the art molecular, genomic, computational and imaging approaches, in order to reach an integrated understanding of these different levels of genome organization.
Role of Polycomb Group Proteins in genome regulation
Polycomb group (PcG) and trithorax group (trxG) proteins are key regulators of the expression of major developmental genes. PcG proteins are able to silence gene expression, while trxG proteins counteract gene silencing in the appropriate cells. The current model to explain PcG recruitment in Drosophila, proposes that a sequence-specific DNA binding proteins, the best studied of which is called PHO, bind at so-called Polycomb response elements (PREs). PHO might recruit the PcG complexes PRC1 and PRC2. PRC2 complexes contain the core subunit E(z), a histone methyltransferase that trimethylates histone H3 lysine 27 (H3K27me3), Su(z)12, Esc and Nurf55. H3K27me3 is, in turn, recognized by the chromo domain of the PC subunits of PRC1, which also contains Ph, PSC and Sce/dRing. On the other hand, PRC1 is actually a family with various canonical and variant complexes, that have in common a catalytic activity carried out by the dRing protein, that can ubiquitylate the K119 of histone H2A, a mark that is recognized by a specific subset of the PRC2 complexes. Mammalian PcG components are recruited to DNA unmethylated CpG islands and, once recruited, set up a similar maintenance system. Therefore, this pathway forms a self-sustaining silencing loop, such that once recruited, PcG complexes can propagate silencing through cell division. Genome-wide mapping studies have shown that PcG target genes encode for components controlling major signalling pathways and, importantly, PcG misexpression has also been associated with many cancer types, including breast and prostate cancer.
In addition to their role in cellular memory, PcG proteins participate in dynamic gene regulatory processes. In flies, different cell lines have a partially different set of PcG bound sites and H3K27me3-marked genomic regions change during development. In mammalian embryonic stem cells, many PcG target genes have been reported to bear both repression- and activation-associated marks. Upon differentiation, these “bivalent states” are resolved into fully active or fully repressed. In some instances, PcG components may even activate transcription, although it is unclear how widespread this phenomenon is. Importantly, PcG proteins regulate the organization of their target genes in the three-dimensional space of the nucleus, and this regulatory function is involved in the maintenance of cellular memory.
We would like to understand the molecular mechanisms of action of these factors, the role of regulation of higher order chromatin structure and nuclear organization in gene expression, and the molecular mechanisms at the base of this processes. In particular, our research aims at (1) understanding, on a genome-wide scale, how these proteins are targeted to DNA and what are the consequences of this targeting on chromatin structure; (2) understanding the effect of PcG proteins on cell proliferation, cell differentiation and cell polarity, and dissecting the key components regulated by PcG proteins to modulate these pathways in specific tissues and developmental processes; (3) identifying the rules governing the distribution of their target genes in the cell nucleus and the effect of this organization on gene expression.
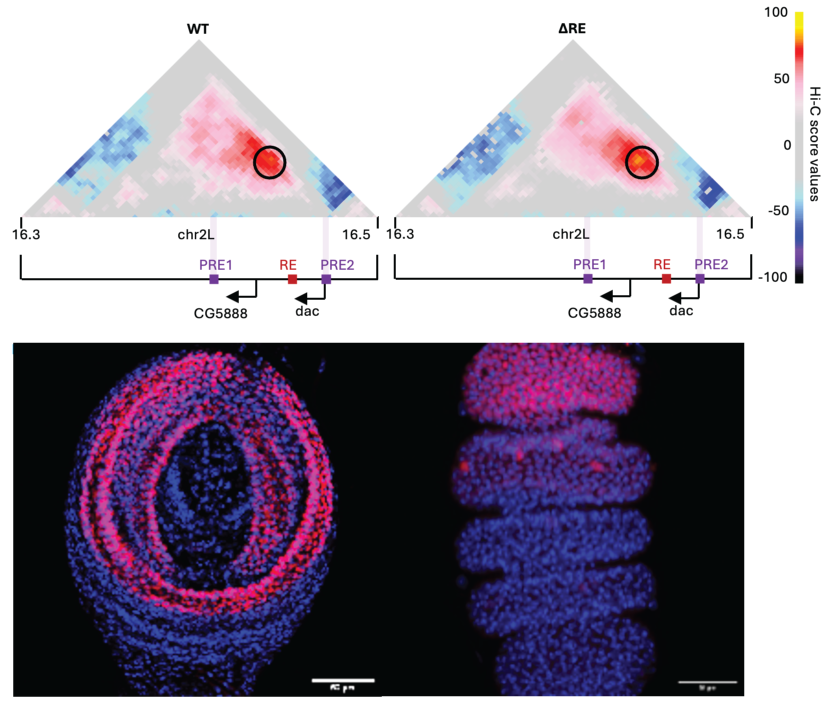
Top: Hi-C score maps of the dachshund (dac) locus in 3rd instar imaginal leg disc in WT (left) or ΔRE (right) Drosophila. A black circle indicates the position of the chromatin loop between the daschund PREs. PREs and and promoter positions are indicated on the bottom. Bottom: DAC immunostaining in WT larval 3rd larval instar (left) or early pupal imaginal leg discs (right). Scale bar= 30 μm.
WaddingtonMemory: Deciphering the role of regulatory factors driving epigenetic inheritance of alternative chromatin states
Epigenetics, the study of molecules and mechanisms that perpetuate alternative gene activity states in the context of the same DNA sequence, is an exciting field with important epistemological and biomedical implications, but the molecular mechanisms underlying epigenetic inheritance are still little understood. Polycomb group proteins are pleiotropic chromatin components that have been suggested to be capable of driving epigenetic inheritance and their dysregulation leads to cell fate changes and is associated with cancer. PcG misexpression has also been associated with many cancer types, including breast, prostate and hematological malignancies. The data suggest that the role of Polycomb components vary in different cancer types and there is a great interest in understanding the molecular mechanisms at play.
We and others have shown that mutations affecting PRC1 subunits trigger neoplastic tumours in Drosophila , suggesting that PRC1 components act as neoplastic tumor suppressors in flies. Strikingly, we discovered that a transient decrease in expression of a Polycomb gene can drive the formation of tumors of epigenetic nature, i.e. in the absence of DNA mutations. The goal of the WaddingtonMemory project is to decipher how epigenetic components can lead to stable changes in cell fate.
Aim 1: Identify the molecular steps leading to epigenetic cell fate derailment and to cancer development following transient Polycomb protein depletion in Drosophila. We will perform a time‑course study using bulk and single‑cell multiomic and imaging approaches in order to dissect the dynamics of cell fate transformation.
Aim 2: Identify the Polycomb‑targets leading to cell fate dysregulation and decipher their mechanistic role. We will test candidate factors identified in Aim 1 in order to identify those that drive cell fate derailment and to elucidate their mode of action.
Aim 3: Test the role of epigenetic inheritance in mouse models for cell differentiation, including gastruloids, an in vitro system that reflects cell differentiation events typically found in early embryogenesis.
Together, this groundbreaking project will reveal how epigenetic components drive cell fate derailment and it will establish robust paradigms that can be utilized by the scientific community to discriminate between epigenetic inheritance and DNA sequence‑mediated cell transformation.
The WaddingtonMemory project is and Advanced Investigator Grant funded by the European Research Council
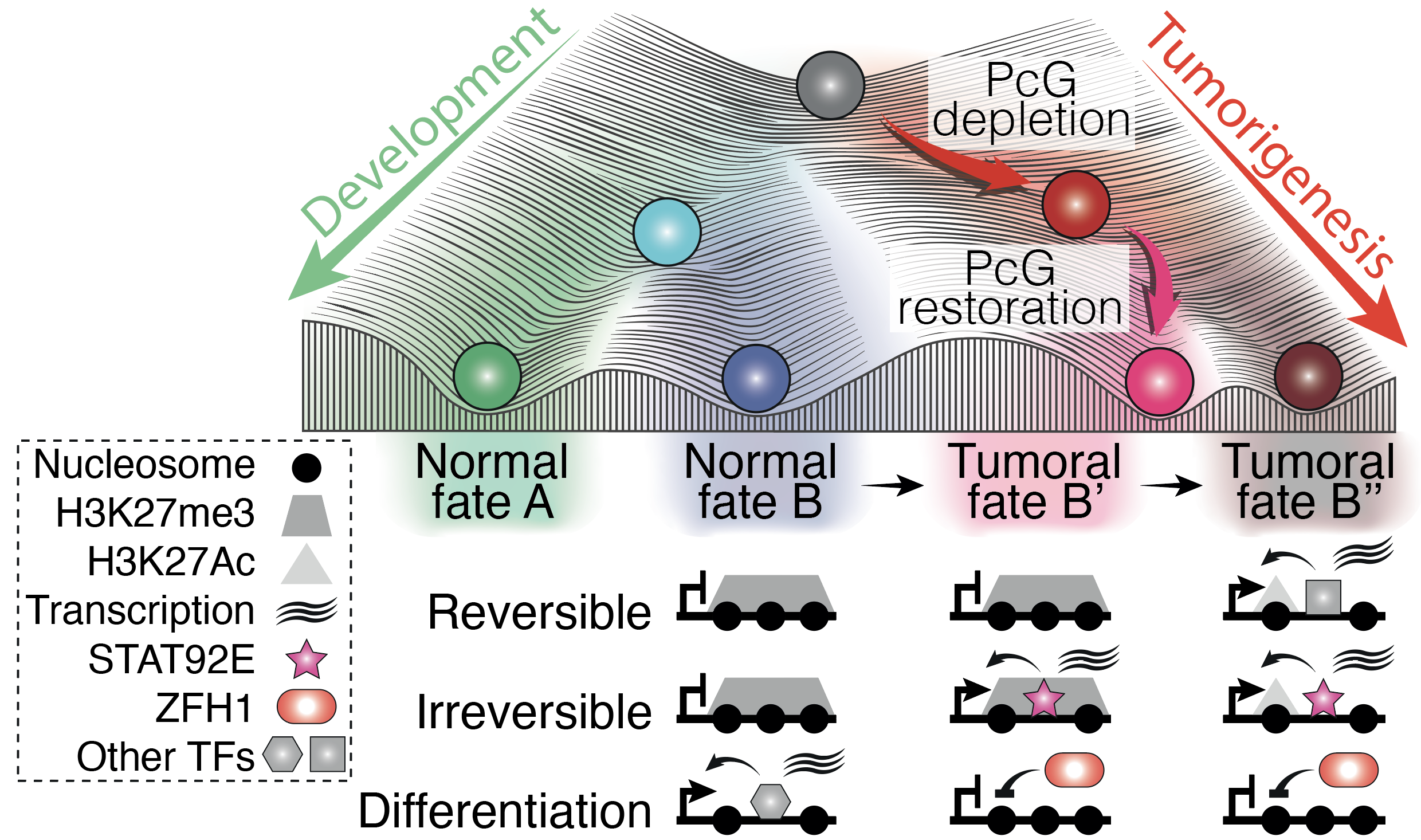
Transient PcG perturbation triggers an irreversible switch to a cancer cell fate. Resulting cancers maintain persistent transcriptional changes supported by the STAT92E activator and the ZFH1 repressor.
Methods used in our lab
Our lab has interdisciplinary expertise and exploits multiple experimental and analytical cutting-edge approaches as well as model organisms or cell systems. We work with Drosophila, an invaluable model organisms with powerful genetic tools that allow us to perform in vivo studies. Drosophila results are also relevant for mammalian biology because of the extensive similarities from flies to humans in terms of regulatory logic of genome, cell and developmental regulation. As a complementary system we use Embryonic Stem Cells (ESCs), a powerful experimental model for the study of pluripotency and cell differentiation toward multiple lineages. Specific differentiation protocols have been established in order to mimic pre- or post-implantation embryonic development and we are using them to decipher the role of epigenetic inheritance in different cell lineages.
To test mechanistic hypotheses in vivo, we exploit elaborated molecular and developmental genetic approaches in flies that allow us to deplete or ectopically express any component of interest in a cell type, tissue or temporal stage of choice. Similarly, we use CRISPR technology and reversible overexpression or protein depletion systems in cell culture to analyse the role of specific components in mammalian genome regulation and cell differentiation.
We use state-of-the art genomic and epigenomic techniques to map the epigenome, transcriptome and the proteome of the cells of interest. We are currently developing low-cell, single-cell or single-nucleus multiomic methods to enable the analysis of low cell numbers or of individual cells, with the aim of understanding genome structure and function. In particular, we have strong expertise in 3D genome architecture mapping using Hi-C and microC methods and their derivatives. We also use advanced imaging, either in fixed tissues or in live applications. We develop multiplex microscopy techniques to study proteins, RNAs and the 3D folding of chromatin, individually or simulateously.
Furthermore, we perform sophisticated computational analysis of chromatin, 3D genome and transcriptome as well as mathematical modelling of chromosome folding and gene expression regulation in order to reach a quantitative understanding and to be able to predict the response of our model systems to regulatory changes or external stimuli.
Finally, we constantly strive to improve existing methods and look forward to develop or adopt new approaches or techniques.